Histiocitosis de células de Langerhans: una rara enfermedad del sistema inmune
Las células de Langerhans son células dendríticas que actúan como presentadoras de antígenos, jugando un papel fundamental en el inicio y regulación de la respuesta inmune.
Normalmente se encuentran en todo el cuerpo, especialmente en la piel, desde la cual migra, para presentar a los linfocitos T ante aquellos antígenos que han logrado penetrar la barrera de la piel e iniciar la respuesta inmune específica.
Estas células pueden sufrir un trastorno poco frecuente y comenzar a proliferar de manera descontrolada, causando una patología conocida como histiocitosis de células de Langerhans (HCL). Los histiocitos se empiezan a acumular en el cuerpo, con la subsiguiente infiltración y afectación de órganos.
En esta neoplasia, las células de Langerhans inmaduras- que se encuentran en exceso- generalmente forman tumores llamados granulomas, por lo que varios investigadores la consideran una forma de cáncer, pero esta clasificación es controvertida.
Anteriormente, se usaban distintos nombres para las variadas formas clínicas de histiocitosis de células de Langerhans, que incluían granuloma eosinofílico- que aún se mantiene- enfermedad de Hand-Schüller-Christian y enfermedad de Letterer-Siwe.
La histiocitosis de células de Langerhans es un trastorno cuya prevalencia se estima en 1 a 2 en 100,000 personas. Aunque la enfermedad puede presentarse a cualquier edad, la edad máxima en el momento del diagnóstico es entre 1 y 3 años. La mayoría de los afectados son niños, con una incidencia es de 5 a 8 casos/millón de niños.
Manifestaciones clínicas
Los signos y síntomas de la HCL varían ampliamente según los órganos que han sido infiltrados.
Los pacientes se dividen en grupos basándose en el compromiso de los órganos:
- Sistema único: La enfermedad de un solo sistema es la afectación unifocal o multifocal de uno de los siguientes órganos: hueso, piel, ganglios linfáticos, pulmones, sistema nervioso central u otros sitios inusuales (p. ej., tiroides, timo).
- Compromiso multisistémico: Este trastorno afecta uno o varios sistemas de órganos, con distintos grados de riesgo, según el órgano o sistema implicado y el compromiso celular infiltrativo.
Los hallazgos clínicos van desde una lesión osteolítica aislada que se resuelve espontáneamente, hasta un síndrome similar al linfoma con falla multiorgánica fatal, sin presentar evidencia celular de malignidad.
En aproximadamente el 80% de las personas afectadas, uno o más granulomas se desarrollan en los huesos, causando dolor e inflamación. Aquellos que ocurren en el cráneo o en los huesos largos de los brazos o las piernas pueden causar una fractura.
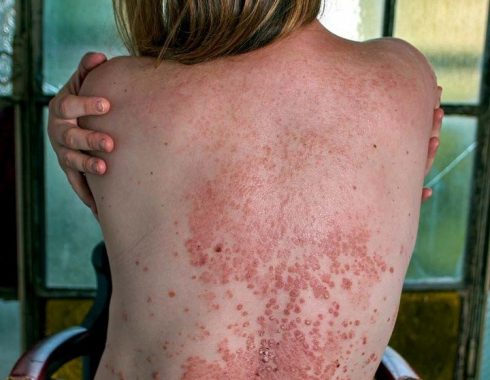
Los granulomas también aparecen con frecuencia en la piel, evidenciándose como lesiones ampollares, protuberancias rojizas o erupciones que pueden ser diversa gravedad.
La hipófisis también puede verse infiltrada, alterando así la síntesis de hormonas de las que es responsable, por lo que, sin una suplementación hormonal, las personas afectadas pueden experimentar pubertad retrasada o infertilidad. Además, el daño de esta glándula puede generar una diabetes insípida y la disfunción de la tiroides, con los correspondientes cuadros clínicos asociados.
En 15 a 20% de los casos, la histiocitosis de células de Langerhans afecta los pulmones o el hígado. En caso que afecte a los órganos hematopoyéticos (formadores de células sanguíneas), puede ser mortal, porque conduce a una reducción general en el número de células sanguíneas (pancitopenia), con riesgos de anemia, infecciones y hemorragias internas graves.
La afectación pulmonar, que aparece como una inflamación de los bronquiolos y los vasos sanguíneos pulmonares, provoca rigidez del tejido pulmonar, problemas respiratorios e incremento del riesgo de infección.
Otros signos y síntomas que pueden ocurrir en la HCL, dependiendo de qué órganos y tejidos estén infiltrados por células de Langerhans, incluyen ganglios linfáticos inflamados, dolor abdominal, ictericia, pubertad tardía, exoftalmia (ojos saltones), mareos, irritabilidad y convulsiones. Aproximadamente 1 de cada 50 personas afectadas experimentan algún grado de deterioro de la función neurológica.
Antecedentes genéticos
La histiocitosis de células de Langerhans generalmente no se hereda y habitualmente ocurre en personas sin antecedentes de la patología en su familia.
Si bien se han identificado algunas familias con varios casos de histiocitosis de células de Langerhans, se desconoce el patrón de herencia.
Como hallazgo, en aproximadamente la mitad de los enfermos con HCL se han identificado mutaciones somáticas en el gen BRAF en las células de Langerhans, las cuales se adquieren durante la vida de una persona y están presentes solo en ciertas células, por lo que no son heredables.
El gen BRAF codifica las instrucciones para sintetizar una proteína que en condiciones normales se activa y desactiva en respuesta a señales que controlan el crecimiento y el desarrollo celular. Pero al existir esta mutación, la proteína BRAF esté continuamente activa en las células afectadas, favoreciendo el crecimiento y división descontrolada de las células de Langerhans.
También se han identificado mutaciones en otros genes en las células de Langerhans de algunos individuos con HCL, por lo que algunos investigadores creen que otros factores, como las infecciones virales y las toxinas ambientales, también pueden influir en el desarrollo de este trastorno.
Diagnóstico
La histiocitosis de células de Langerhans a menudo se diagnostica en la infancia, pero puede aparecer a cualquier edad.
La mayoría de las personas con esta patología de inicio en el adulto son fumadores actuales o pasados, y en aproximadamente dos tercios de los casos de aparición en este grupo etario el trastorno afecta solo a los pulmones.
La gravedad de la histiocitosis de células de Langerhans, así como sus signos y síntomas, varía ampliamente entre los individuos afectados.
El diagnóstico se realiza en base a una biopsia, ante la sospecha de una HCL en pacientes, especialmente si son jóvenes, y presentan infiltrados pulmonares sin causa reconocida, lesiones óseas o anomalías oculares o que afectan cara y cráneo. También está indicada en el caso de los niños pequeños con erupciones características o enfermedad multiorgánica grave sin causa aparente.
Además se realizan estudios de laboratorio e imagenología para ver el alcance del compromiso orgánico.
Tratamiento
En muchos pacientes con histiocitosis de células de Langerhans el trastorno puede desaparecer con el tratamiento adecuado. Incluso puede resolverse de manera espontánea, especialmente si afecta solo a la piel.
Sin embargo, algunas complicaciones asociadas, como la diabetes insípida u otros daños tisulares y orgánicos, pueden ser permanentes, por lo que pueden requerir:
- Tratamiento de sostén.
- Terapias de reemplazo hormonal en caso de hipopituitarismo, principalmente en diabetes insípida.
- Quimioterapia cuando existe infiltración multisistémica, afectación multifocal de un solo sistema y daño de ciertas zonas anatómicas, como en el caso de lesiones craneales
- En ocasiones se requiere cirugía, inyección de corticosteroides o, rara vez, radioterapia (por lo general, en caso de compromiso óseo unifocal).
Pronóstico
El pronóstico y la morbimortalidad depende de la edad y del grado de afectación- si es unifocal o multisistémico- siendo bueno en los pacientes con HCL limitada a la piel, los ganglios linfáticos o los huesos. En cambio, ante el compromiso de los órganos en riesgo- como son sistema hematopoyético, hígado o bazo- el pronóstico empeora.
Con tratamiento, la tasa de supervivencia global de los pacientes con enfermedad multisistémica sin riesgo de compromiso orgánico es del 100%, pero la supervivencia libre de eventos asociados a la patología es de alrededor de 70%.
La muerte es poco frecuente en pacientes con compromiso orgánico que no responden al tratamiento inicial y la recidiva de la enfermedad es habitual. Además, puede observarse una evolución crónica de la enfermedad, con exacerbaciones y remisiones, especialmente entre los adultos.
Algunos hallazgos sugieren que los pacientes que tienen las mutaciones BARFV600E son más propensos a sufrir recaídas, pero aún queda mucho por investigar y aprender sobre esta rara enfermedad.