Qué es la Hemocromatosis hereditaria, la enfermedad en que el hierro pasa a ser tóxico

La hemocromatosis corresponde a un grupo de patologías ocasionadas por un nivel excesivo de hierro en el organismo, debido a una acumulación de este mineral, que puede ser hereditaria o bien adquirida, secundaria a múltiples transfusiones sanguíneas, hepatopatías, alcoholismo crónico u otras patologías relacionadas con el metabolismo del hierro.
La hemocromatosis hereditaria (HH) es una enfermedad con base genética, en la que se produce una excesiva absorción intestinal del hierro procedente de la dieta, que termina acumulándose en el parénquima de diversos órganos.
Clínicamente se presenta con cirrosis, diabetes mellitus, artropatía, miocardiopatía, hiperpigmentación cutánea, hipogonadismo, y aumento del riesgo a padecer cáncer hepático.
La HH representa la hepatopatía hereditaria metabólica más frecuente. La mutación que la origina, ocurre especialmente en los habitantes de las regiones de origen céltico, siendo muy rara en poblaciones orientales como indios y chinos.
Según estadísticas de la OMS, afecta a una de cada 200- 300 personas, con una mayor prevalencia en los hombres con respecto a las mujeres, quienes presentan pérdidas mensuales de hierro a través de la menstruación.
En el caso de los hombres, la HE aparece especialmente entre los 30 y los 50 años de edad, mientras que en las mujeres, la sintomatología se hace evidentes después de la menopausia, aunque también se puede manifestar en pacientes más jóvenes.
Patogénesis
El gen HFE codifica la síntesis de una proteína transmembrana de amplia distribución tisular, que se une al receptor de la transferrina. La mutación C282Y se manifiesta en la ausencia de expresión de la proteína HFE en la superficie celular. Por su parte, la mutación H63D se traduce en la alteración de la estructura terciaria de esta proteína, que da como resultado una absorción excesiva de hierro a través de la mucosa duodenal, con su acumulación progresiva en las células epiteliales del hígado, hipófisis, páncreas y músculo cardíaco, provocando fibrosis e insuficiencia funcional, con todos los signos y síntomas que ello implica.
Dentro de los órganos afectados, el hígado suele ser el más perjudicado por esta sobrecarga de hierro, que puede llevar a una cirrosis que termine en un hepatocarcinoma.
Los pacientes con HE también pueden desarrollar diabetes- debido al depósito de hierro en las células beta del páncreas- además de daño cardíaco y artropatía.
Tipos de hemocromatosis hereditaria (HH)
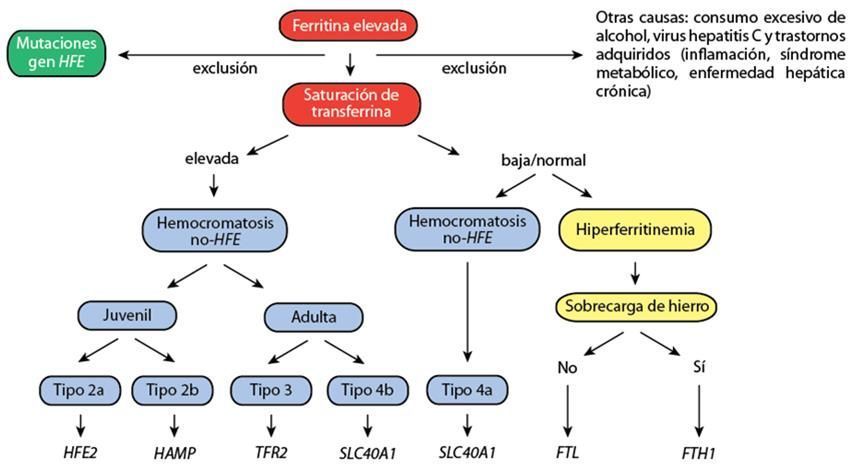
Actualmente se distinguen 5 tipos de hemocromatosis en función de la causa genética. La mayoría de subtipos de HE son enfermedades raras que afectan a menos de 1/1,000,000 de individuos.
1. Hemocromatosis Hereditaria tipo 1 (HH tipo 1)
La hemocromatosis de tipo 1 o hemocromatosis clásica tiene la mayor prevalencia, presentándose en 1 de cada 200 personas, en Occidente. Suele ser más frecuente en varones de raza blanca, de origen caucásico, y de más de 50 años.
Presenta una herencia autosómica recesiva. Se debe a mutaciones en el gen HFE, que se ubica en el cromosoma 6p22.2.
La mutación C282Y en homocigosis predispone a padecer esta patología, sin embargo, como esta mutación se caracteriza por presentar una penetrancia incompleta, no todas las personas con este genotipo desarrollarán HE.
Aparte de la mutación C282Y, se han encontrado otras mutaciones puntuales más raras y poco frecuentes del gen HFE, detectadas solo al realizar la secuenciación del gen completo.
2. Hemocromatosis Hereditaria tipo 2 (HH tipo 2)
Este tipo de HH es la forma más temprana y grave de la enfermedad, aunque es muy infrecuente.
La sintomatología suele aparecer antes de los 25 años de edad. El cuadro clínico muestra el predominio de hipogonadismo y síntomas cardiacos- en comparación a la enfermedad hepática- además de la mayor gravedad de los síntomas, siendo frecuente en estos casos la muerte por insuficiencia cardiaca antes de la cuarta década de vida.
La HH tipo 2 es de carácter autosómico recesivo y se debe a mutaciones en dos genes:
- El gen HFE2se localiza en el cromosoma 1q21.1 y codifica para la proteína hemojuvelina, que está presente en la mitad de los casos.
- El gen HAMPse encuentra en el cromosoma 19q13.12. Estas mutaciones son menos frecuentes.
3. Hemocromatosis Hereditaria tipo 3 (HH tipo 3)
La HH tipo 3 es menos frecuente que la HH tipo 1. La manifestación clínica es similar a la HH tipo 1, pero la afectación suele ser más severa y con presentación a edades más tempranas, con depósitos de hierro predominantemente hepáticos.
Se caracteriza por hiperferritinemia, elevada saturación de transferrina y hierro sérico, que sobrecarga de hierro a varios tejidos, especialmente el hepático.
La HH tipo 3 afecta especialmente a adultos de mediana edad, pero también se han diagnosticado casos en niños, adolescentes y adultos jóvenes.
Este tipo de HH se debe a mutaciones en el gen del receptor 2 de la transferrina (TFR2), localizado en el cromosoma 7q22.1.
4. Hemocromatosis Hereditaria tipo 4 (HH tipo 4)
Esta forma de HH también es llamada enfermedad de la ferroportina, es una forma rara pero más común que la HH tipo 2 y 3.
Su patrón de herencia es autosómico dominante, manifestándose en mutaciones de los genes FTL y SLC40A1.
En la HH tipo 4 existe una temprana acumulación de hierro en las células del sistema reticuloendotelial y un incremento importante de los niveles de ferritina sérica, antes de evidenciarse un aumento en la saturación de transferrina.
5. Hemocromatosis Hereditaria dominante debida a mutaciones en el gen BMP6
Este tipo de HH es de reciente descubrimiento. Los pacientes que se han diagnosticado presentan acúmulos de hierro hepático, con niveles de ferritinas aumentados y valores de saturación de transferrina normales o elevados.
La clínica de estos pacientes es similar a una HH tipo 4. Algunos sujetos presentan factores asociados como sobrepeso y alcoholismo, que agravan las manifestaciones clínicas.
La herencia de este tipo de HH es de carácter autosómico dominante y es debida a mutaciones en el gen BMP6, localizado en el cromosoma 6p24.3. La expresión de este gen se modula por los niveles séricos de hierro, activándose en condiciones de sobrecarga del mineral.
Diagnóstico de HH
La hemocromatosis hereditaria puede ser difícil de diagnosticar, puesto que la causa de la sintomatología temprana, como articulaciones rígidas y fatiga, son manifestaciones inespecíficas, que pueden darse en otras patologías.
Muchas personas que padecen de esta afección no presentan signos ni síntomas, aparte de niveles elevados de hierro sérico.
Su diagnóstico se basa en detectar mediante análisis de laboratorio un aumento en el índice de saturación de la transferrina y en los niveles de ferritina y hierro séricos.
El diagnóstico se confirma a través de una prueba genética que detecta las mutaciones del ADN en el gen HFE, si la persona tienes niveles altos de hierro en la sangre.
Detección de hemocromatosis para personas saludables
Se recomienda realizar análisis genéticos para todos los familiares de primer grado (padres, hermanos e hijos), de cualquier paciente que haya sido diagnosticado con hemocromatosis.
Estudios adicionales
El médico puede sugerir otros estudios para confirmar el diagnóstico y detectar otros problemas, como análisis de función hepática, resonancia magnética nuclear y biopsias hepáticas, para detectar daños tisulares en órganos con posible afectación.
Tratamiento
El principal tratamiento para los pacientes con HH consiste en la realización de flebotomías terapéuticas periódicas para remover el exceso de hierro presente en el organismo. Este tratamiento es muy efectivo, económico y casi carente de efectos secundarios. Los pacientes también pueden tratarse mediante eritroaféresis.